Примерное меню новорожденного с ФКУ 3-х недельного возраста с массой тела 3500гр Толерантность фенилаланина 140 мг/сутки
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 40 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 140 | 8,4 | 17,15 | 47,6 | 378 | |
| Грудное молоко PKU 1- mix (49г + 293 мл воды) = | 312 | 140 | 3.43 | 12.48 | 22.15 | 221.52 |
| 323 | 4.96 | 13.52 | 27.58 | 251.86 | ||
| Суточная норма | 140 | 8,39 | 26 | 49,73 | 473,38 |
Ребёнок может получить
- 3 x грудное молоко в объёме 104 мл и 4 x Milupa PKU 1-mix 94мл (на 1 порцию 12,2г порошка или 2,8 мерки) или
- 7 x 54мл Milupa PKU 1-mix (на 1 порцию 7,7г препарата или 1,8 мерки) и 7 x 44 мл грудного молока
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 40 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 140 | 8,4 | 17,15 | 47,6 | 378 | |
| Грудное молокоXP Analog LCP (38г+228 мл воды) = | 312 | 140 | 3.43 | 12.48 | 22.15 | 221.52 |
| 252 | 4.94 | 8.74 | 20.52 | 180.5 | ||
| Суточная норма | 140 | 8,37 | 21,22 | 42,67 | 402,02 |
Ребёнок может получить
- 3 x грудное молоко в объёме 104 мл и 4 x 63мл XP Analog LCP (на 1 порцию 9,5г порошка или 1,9 мерки) или
- 7 x 36мл XP Analog LCP (на 1 порцию 5,0г препарата или 1,0 мерка) и 7 x 45мл грудного молока
Симптомы
Первые симптомы возникают в возрасте 2–6 месяцев. ребенок становится раздражительным, беспокойным, или, наоборот, его ничего не интересует, он ко всему индифферентный, болезненно спокойный. Иногда болезнь проявляется рвотой.
В 2 года ребенок с фенилкетонурией отстает в психическом развитии: идиотия (60 % детей), легкая олигофрения (10 %). Рост нормальный либо немного меньше нормы. Малыш начинает позже сидеть и ходить, у него позже, чем у сверстников, появляются зубы, может наблюдаться уменьшение черепа.
Дети, страдающие этой болезнью, имеют особенную внешность: светлые волосы, голубые глаза, лишенная пигмента кожа. Для них характерен так называемый “мышиный” запах, излишняя потливость, синюшность конечностей. Определить ребенка с ФКУ можно и по поведению: у него своеобразная походка – мелкие шаги, опущенные плечи и голова, легкие покачивания в разные стороны – и поза – ноги согнуты в тазобедренных и коленных суставах и широко расставлены. Сидит кроха, поджав ноги под себя, что обусловлено повышенным мышечным тонусом.
Дети с фенилкетонурией имеют сильную чувствительность к солнечным лучам и страдают патологическими травмами. У них определяют артериальную гипотонию, экзему и дерматит. У некоторых больных наблюдаются эпилептические припадки, которые с возрастом проходят.
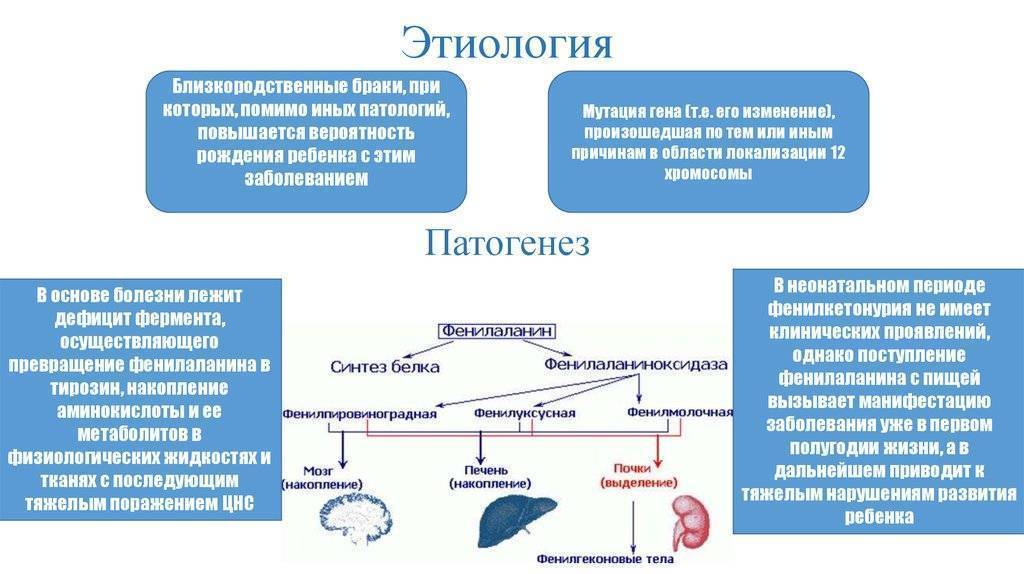
Этиология и патогенез
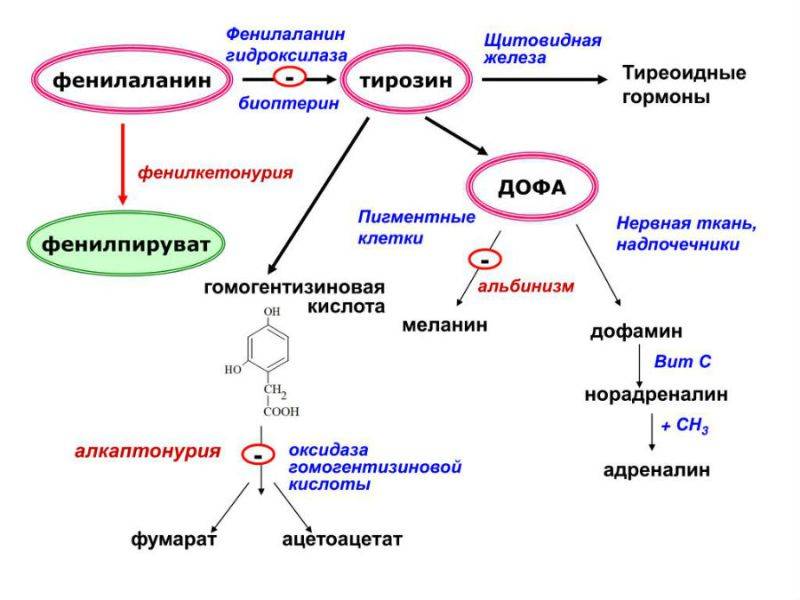
Наследуется по аутосомно-рецессивному типу (см. Наследование). В основе биохимических нарушений лежит отсутствие вырабатываемого печенью фермента фенилаланин-4-гидроксилазы, что приводит к блокированию превращения фенилаланина (см.) в тирозин (см.). При этом концентрация фенилаланина в крови повышается до 15—20 мг/100 мл и более (норма до 2,5 мг/100 мл). В результате дезаминирования из фенилаланина образуется фенилуксусная, фенилмолочная, фенилпировиноградная кислоты, а также фенилацетилглутамин, выделяющиеся в избытке с мочой. Вторично нарушается обмен тирозина и триптофана (см.), что приводит к недостаточному образованию норадреналина (см.), адреналина (см.), дофамина (см. Катехоламины), меланина (см.). Описанные нарушения возникают в первые дни после рождения ребенка, достигают максимума на 1—2-й неделе и сохраняются при отсутствии лечения на протяжении всей жизни больного.
Диетотерапия
Именно лечебная диета максимально эффективна в предотвращении интеллектуального дефицита при тяжелой (классической) ФКУ. Наибольшее значение имеет возраст пациента к моменту начала диетотерапии ( снижается примерно на 4 балла за каждый месяц от рождения до начала лечения). Подходы к диетотерапии ФКУ в разных странах несколько различаются, но сами их принципы являются согласованными.
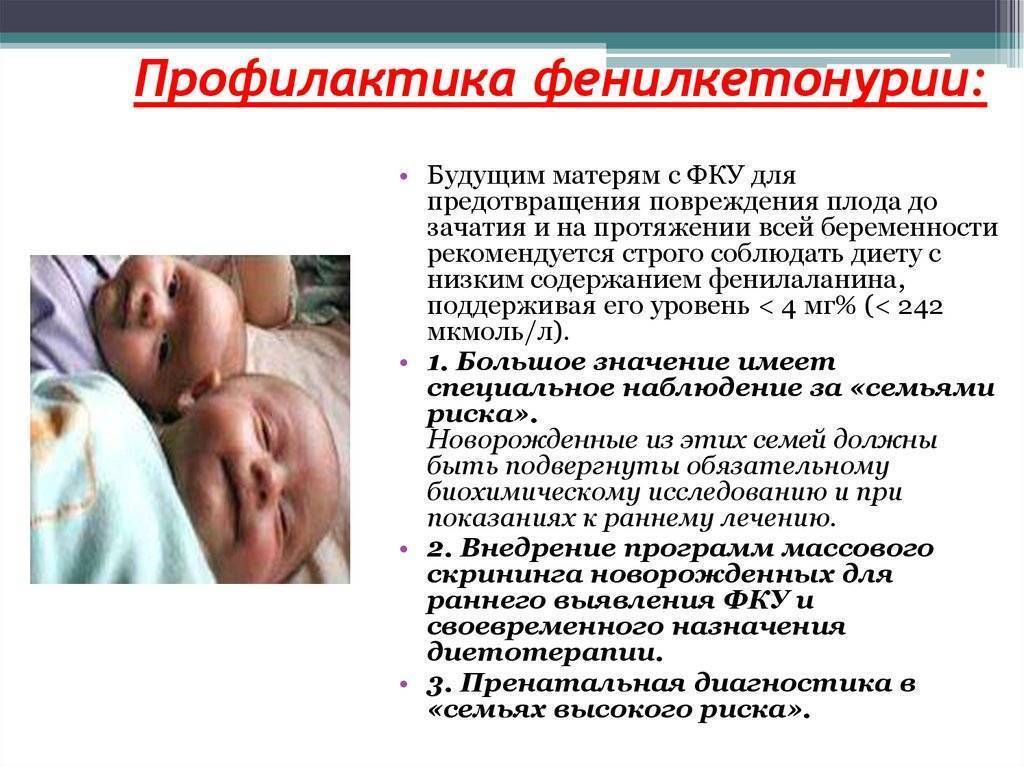
Диетические ограничения не показаны младенцам, у которых уровень фенилаланина в крови находится в пределах 2–6 мг% (120–360 мкмоль/л). Основа диеты при ФКУ — назначение рационов питания с низким содержанием фенилаланина, источником которого является белковая пища. Такая диета назначается всем пациентам первого года жизни. Она должна быть назначена детям с выявленной ФКУ до 8-недельного возраста; ее применение в более позднем возрасте гораздо менее эффективно .
Диета ФКУ в практике
Грудное молоко является самым лучшим питанием для младенца, содержит все питательные компоненты необходимые для нормального роста и развития ребёнка. Диета грудного ребёнка с ФКУ базируется на смесях (препаратах) без фенилаланина, которые являются главными источниками белка, витамин, микро- и макроэлементов. Грудное молоко и молочная смесь для младенцев дополняют эту диету необходимым для роста фенилаланином. Количество препарата ФКУ, грудного молока или молочных смесей надо систематически изменять в зависимости от индивидуальной толерантности фенилаланина, а также и от потребностей растущего организма. В течение первого года жизни толерантность фенилаланина быстро изменяется постоянно уменьшаясь, в связи с этим концентрацию фенилаланина в крови необходимо контролировать в определённых промежутках времени, а диету модифицировать. Все компоненты необходимые для приготовления пищи должны быть с точностью взвешены или отмерены.
Способ приготовления смесей:
Во время приготовления смесей следует сохранять правила гигиены. Бутылки, соски, крышки и остальные необходимые для использования приборы перед каждым использованием необходимо вымыть и стерилизовать в кипящей воде в течении 5 минут.Свежую воду кипятить в течении 5 минут. Охладить до температуры 40° C. Стандартный раствор получаем растворяя 1 плоскую мерную ложечку, находящуюся в упаковке, порошка в 30 мл воды.
3 мерки порошка + 90 мл воды = 100 мл
Соответствующее количество воды влить в бутылку, добавить отмеренное количество порошка, закрутить, встряхнуть бутылку, полностью смешивая содержимое. Перед кормлением следует проверить на тыльной стороне ладони температуру смеси (должна быть тёплой).
Меню для грудного ребенка с ФКУ
в возрасте 2 месяцев с массой тела 5 000г, с индивидуальной толерантностью фенилаланина 175мг/сутки
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 35 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 175 | 12 | 24 | 68 | 540 | |
| Грудное молокоXP Analog LCP 60г + 360мл воды | 390 | 175 | 4.29 | 15.6 | 27.3 | 276.9 |
| 420 | 6.06 | 16.56 | 33.78 | 308.4 | ||
| Суточная норма | 175 | 12,09 | 29,4 | 59,7 | 561,9 |
* Ребёнок может получить:
- 1 способ:
- 3 x 130мл грудного молока и 3 x 120мл XP Analog LCP (1 порция = 20г препарата XP Analog LCP = 4 мерки + 120мл воды)
- 2 способ:
- 6 x 60мл XP Analog LCP (1 порция = 10gг препарата XP Analog LCP = 2 мерки + 60мл воды) и 6 x 60мл грудного молока
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 35 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 175 | 12 | 24 | 68 | 540 | |
| Грудное молокоMilupa PKU 1-mix (60г + 418мл воды) = | 390 | 175 | 4.29 | 15.6 | 27.3 | 276.9 |
| 420 | 6.06 | 16.56 | 33.78 | 308.4 | ||
| Суточная норма | 175 | 10,35 | 32,16 | 61,08 | 584,4 |
* Ребёнок может получить:
- 1 способ:
- 3 x 130мл грудного молока и 4 x 104мл Milupa PKU 1-mix (1 порция = ???г препарата Milupa PKU 1-mix = 3,5 мерки + 104 мл воды)
- 2способ:
- 7 x 60мл Milupa PKU 1-mix (1 порция = 8,6г препарата Milupa PKU 1-mix = 2 мерки + 60мл воды) и 7 x 56мл грудного молока
Как диагностировать болезнь
Синдром Дауна можно распознать еще в пренатальном периоде, то есть во время беременности. Что касается его мозаичной формы, то в отдельных случаях его диагностика на данном этапе затрудняется ввиду скудности проявлений и поражении не всех хромосом. В таком случае его раскрывают только после родов.
Выявить заболевание удается несколькими методами:
1.УЗИ-диагностика. Ее проводят каждые 3 месяца беременности. Уже в первом триместре, с 8 по 12 неделю, можно распознать некоторые признаки СД. Например, утолщение воротниковой зоны.








2.Во втором триместре УЗИ покажет дефекты развития ЦНС, органов пищеварения, выделительной системы и слуха, а также сердечные пороки. На третьем УЗИ специалист увидит мелкие аномалии развития.
3.Анализ крови матери. Первый триместр (с 8 по 12 недели) определяет количество веществ, которые продуцирует плод:
- ассоциированный с беременностью плазменный протеин А (РАРР-А);
- хориогонадотропин человека (ХГЧ). Если уровень ХГЧ повышен, а РАРР-А понижен, это может говорить о патологии;
4.Второй триместр (с 18 по 21 неделю) определяет следующие показатели:
- альфа-фетопротеин;
- ХГЧ;
- свободный эстриол.
Подобные исследования не дают 100% гарантии наличия или отсутствия патологии. Если беременная попала в зону риска, то есть перечисленные анализы показали нестандартный результат, женщине проводят инвазивные процедуры. Они позволяют получить материал плода для уточнения диагноза:
- биопсия хориона – процедура дает возможность получить тканевые образцы плода. Ее осуществляют с помощью пункции брюшной стенки, а также через шейку матки специальными щипцами или катетером. Проводят на ранних сроках беременности, в 8—12 недель;
- амниоцентез – осуществляют в период 14—18 недель. Делают пункцию оболочки плода для забора околоплодных вод;
- кордоцентез проводится на поздних сроках вынашивания. Производят забор крови из пуповины.
Данные инвазивные методы являются довольно опасными, так как существует риск разрыва околоплодного пузыря, выкидыша, а также инфицирования матери и ребенка.
Что такое Фенилкетонурия (ФКУ) у детей –
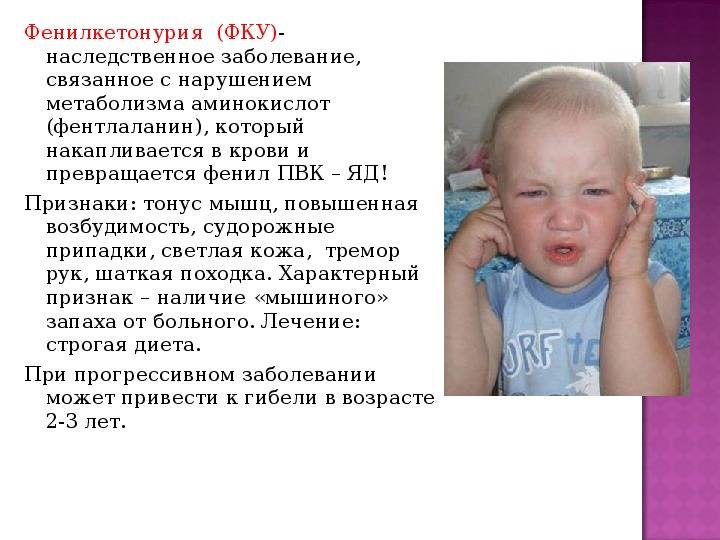
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
Название формы | Уровень фенилаланина |
классическая | выше 20 мг% (1200 мкмоль/л) |
средняя | 10,1–20 мг% (600–1200 мкмоль/л) |
легкая | до 8 мг% (480 мкмоль/л) |
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.










ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Ассортимент продуктов для детей с ФКУ
Ассортимент продуктов для детей с ФКУ существенно ограничен. В периоде максимально строгого соблюдения диеты (грудной и ранний детский возраст) обязательно использование специализированных лечебных продуктов. Целью их применения при ФКУ является замещение источников белка при полном соответствии нормам потребления основных нутриентов детьми (с учетом возраста и конкретной клинической ситуации). Часть лечебных продуктов содержит полиненасыщенные жирные кислоты (омега-6 и омега-3) в соотношении 5:1–10:1; таким источникам пищи отдается предпочтение. Из специальных продуктов применяются сухие аминокислотные смеси, лишенные содержания фенилаланина, с дотацией белкового эквивалента — его артифициального аналога (в количествах, соответствующих возрасту больных ФКУ). Другие доступные в РФ малобелковые продукты для диетотерапии ФКУ включают саго, специальный хлеб, вермишель и другие виды лечебного питания. Эти лечебные продукты (амилофены) основаны на крахмалах, не содержащих трудноусвояемых углеводов и минеральных веществ. Они представлены макаронными изделиями, крупами, саго, специальной мукой, хлебобулочными изделиями, инстантами для приготовления киселей, муссов и т. д. Витаминные добавки повышают питательную ценность продуктов с низким содержанием белков. Существуют также малобелковые продукты зарубежного производства, (Нидерланды-Великобритания), на основе крахмалов (пшеничного, рисового, картофельного, кукурузного и т. д.), включающие макаронные изделия, крупы для приготовления каш, специальные сорта хлеба (из тапиоки, пшеничного и рисового крахмала), печенье, крекеры, сухари, а также мука, различные десерты, приправы и соусы с привлекательным вкусом, значительный ассортимент напитков (включая заменители молока, сливок и кофе) и т. д.. |
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Трудности в соблюдении диеты
Недобросовестное выполнение диетических рекомендаций со стороны родителей:
– Необходимость регулярность приготовления аминокислотной смеси (формулы) – 3-4 дозы в сутки
– Взвешивание продуктов
– Записывание…
– Пересчёты (фенилаланин, белок, калории)
– цена, вкус, запах специализированных продуктов
Психологические трудности детей и подростков:
– Встречи со сверстниками – говорить про ФКУ? Не говорить?
– Желание ничем не отличятся от других
– Желание попробовать „неизвестного”
– Страх перед исключением из групы
– Школьные экскурсии – как незаметно выпить аминокислотную смесь…










– Питание в общественных местах – столовые, рестораны
– Путешествование (необходимость иметь формулу всегда с собой)
Поэтому многие больные не собюдают, или соблюдают неполностью, диетические указания лечящих врачей и диетологов. Чаще всего от диеты отказываются молодёжь и взрослые.
2002 год – исследования (3 английских и 1 австралийский центр)
– у 30 % детей < 4 лет жизни
– почти у 80 % молодёжи в возрасте 15 – 19 лет
уровни ФА были выше допускаемой нормы для возраста!
(Walter et al.2002) JIMD (2010) 33: 665-667 The reality of dietary compliance in the management of phenylketonuria. Anita MacDonald, Hulya Gokmen-Ozel, Margreet van Rijn and Peter Burgard
В Германии – 42 из 89 больных (47 %) было в состоянии соблюдать тщательно диету в период первых 9 лет жизни. (Burgard et al. 1996) JIMD (2010) 33: 665-667 The reality of dietary compliance in the management of phenylketonuria. Anita MacDonald, Hulya Gokmen-Ozel, Margreet van Rijn and Peter Burgard
Международные исследования (10 опытных по лечению ФКУ Европейских центров – том числе Англия, Германия, Польша и др.)
– 1921 больных, период 2007 – 2008 год (1 год)
Результаты:
хороший контроль диеты → уровни ФА в пределах нормы для возраста составляли соответсвенно:
– 88 % – дети < 1 гж
– 74 % – дети в возрасте 1 – 10 лет
– 89 % – дети и молодёж в возрасте 11 – 16 лет
– 65 % – взрослых ( Gokmen-Ozel et al 2009)
Соблюдание диеты среди Европейской молодёжи улучшается.
JIMD (2010) 33: 665-667 The reality of dietary compliance in the management of phenylketonuria. Anita MacDonald, Hulya Gokmen-Ozel, Margreet van Rijn and Peter Burgard
Фенилкетонурия: механизм заболевания
Один из ингредиентов нашей еды — белки. Белки, как растительные (например, в картофеле, рисе, маргарине), так и животные (мясо, рыба, яйца), состоят из мелких частиц, похожих на кирпичи в стене. Затем во время пищеварения эти белки расщепляются на различные строительные блоки, из которых тело строит необходимые структуры, например, мышцы или гормоны.
У пациентов с фенилкетонурией отсутствует фермент, ответственный за преобразование одного из этих строительных блоков: фенилаланина. В результате этого нарушения фенилаланин не может быть превращен в другой строительный блок, тирозин.








В результате фенилаланин и продукты его аномального метаболизма начинают накапливаться в крови ребенка, страдающего фенилкетонурией. В организме тирозин необходим для образования гормонов щитовидной железы (тироксин), гормонов надпочечников (адреналин, норадреналин), для производства красителей (меланинов — это пигмент кожи человека) и для построения других белков организма. У ребенка с фенилкетонурией уровень фенилаланина в крови повышается, а количество тирозина недостаточно для правильного функционирования организма.
Метаболизм фенилаланина
Организм находит другой способ избавиться от избытка фенилаланина — вместо тирозина он превращается в такие соединения, как фенилпировиноградная кислота, фенилмолочная кислота и гидроксифенилуксусная кислота. Эти кислоты тоже являются кетонами, отсюда и название болезни — фенилкетонурия.
Они накапливаются в крови, а затем появляются в моче. Почти в 98% случаев дефект связан с мутацией в гене, кодирующем фермент фенилаланингидроксилазу (ПАГ), а в остальных случаях возникают мутации в генах, кодирующих ферменты, связанные с биосинтезом или метаболизмом кофактора реакции превращения фенилаланина в тирозин.
В Европе заболеваемость фенилкетонурией в среднем составляет 1 из 7 тысяч. живорождений. Это генетическое заболевание, переданное ребенку обоими родителями.
Заболевание вызвано мутацией генов на 12-й хромосоме, которые кодируют ферменты в системе гидроксилирования фенилаланина. Выявлено около 800 различных мутаций в генах, кодирующих фенилаланингидроксилазу (PAH), GTP-циклогидролазу I (GTPCH), 6-пирувилтетрагидробиоптеринсинтазу (PTS) и тетрагидробиоптеринредуктазу (DHPR). – мутации этих генов связаны с заболеванием. Самые распространенные из них касаются гена, кодирующего белок PAH.
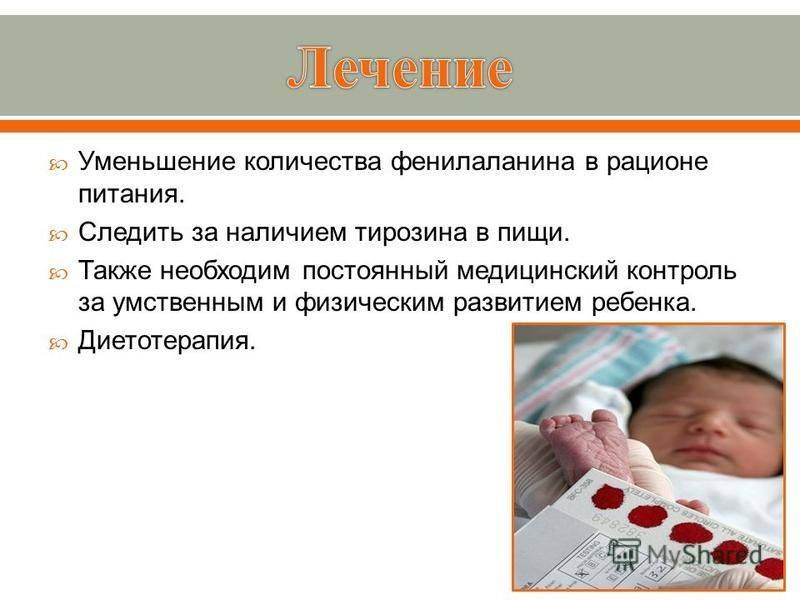
Лечение
Основным способом лечения сегодня является своевременное начало диеты. Необходимо ограничить прием пищевых продуктов, в которых содержится фенилаланин, в частности белковых продуктов. Их заменяют белковыми гидролизатами и аминокислотными смесями. В рационе должны преобладать овощи и фрукты, углеводы, животные и растительные жиры.
Строгая диета соблюдается на протяжении 5 лет. Если все меры соблюдены правильно, то к 12–14 годам ребенок может перейти к обычному питанию. Медикаментозное лечение является периодическим и направлено, прежде всего, на устранение судорог и повышение интеллектуальной деятельности ребенка. Кроме того, дети с ФКУ обязательно проходят курс массажа и лечебной физкультуры, а также посещают специальные занятия для развития логического мышления.
Контролировать содержание аминокислоты в организме необходимо и по окончании диеты. Ее должно быть достаточно для нормального развития и роста ребенка, но при этом она не должна накапливаться в тканях.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Общее описание
Фенилкетонурия (ФКУ) — это тяжкое наследуемое заболевание, происходящее вследствие неправильного обмена ароматических аминокислот и манифестирующееся олигофренией, задержкой физического развития, локомоторными расстройствами. ФКУ заболевает примерно один из 5-10 тыс. новорожденных. Среди больных преобладают девочки, т.к. младенцы мужского пола практически все погибают во время первого года жизни.
В результате того что при ФКУ не расщепляется аминокислота фенилаланин, поступающая с белковой пищей, в организме накапливаются соединения, обладающие нейротоксическим действием, следствием чего является развитие олигофрении. ФКУ наследственно детерминирована и возникает только при условии, что каждый из родителей передал ребенку патологический ген. Практически такое возможно при близкородственных браках. Считается, что порядка 2% популяции являются носителями такого типа гена, но при этом не имеют признаков заболевания.
Online-консультации врачей
| Консультация аллерголога |
| Консультация общих вопросов |
| Консультация специалиста по лазерной косметологии |
| Консультация онколога-маммолога |
| Консультация пульмонолога |
| Консультация трихолога (лечение волос и кожи головы) |
| Консультация детского психолога |
| Консультация ортопеда-травматолога |
| Консультация эндокринолога |
| Консультация радиолога (диагностика МРТ, КТ) |
| Консультация проктолога |
| Консультация вертебролога |
| Консультация эндоскописта |
| Консультация гастроэнтеролога |
| Консультация гомеопата |
Новости медицины
Футбольные фанаты находятся в смертельной опасности,
31.01.2020
“Умная перчатка” возвращает силу хвата жертвам травм и инсультов,
28.01.2020
Назван легкий способ укрепить здоровье,
20.01.2020
Топ-5 салонов массажа в Киеве по версии Покупон,
15.01.2020
Новости здравоохранения
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Коронавирус атаковал уже более 100 стран, заразились почти 120 000 человек,
11.03.2020
Коронавирус атаковал 79 стран, число жертв приближается к 3200 человек,
04.03.2020
Новый коронавирус атаковал 48 стран мира, число жертв растет,
27.02.2020
Что такое синдром Дауна
В норме хромосомный набор человека содержит 23 пары или 46 хромосом. Но при определенных условиях 21 пара способна мутировать (чаще утраиваться), приводя к генетическому дефекту. Именно он и называется синдром Дауна.
Первый раз проявления болезни были зафиксированы в 1866 году врачом из Англии Джоном Дауном. Его имя и дало название синдрому. Однако зависимость между заболеванием и хромосомными парами установлена лишь в 50-х годах 20 века.









Википедия трактует данное понятие как геномное нарушение, при котором кариотип содержит 47 хромосом вместо привычных 46. Это происходит потому, что 1 из 23 пар хромосом – 21, вместо двух имеет 3 копии.
Но под данную трактовку подпадает самый распространенный вид дефекта – трисомия 21 пары, развивающаяся во всех клетках организма. Это самый тяжелый вид заболевания с ярко выраженными патологиями.
Другим вариантом синдромаявляется робертсоновская транслокация. Встречается она чуть реже, чем первый вариант. При ней 21 пара не утроена, а, наоборот, ее хромосомы склеены между собой, а общее количество хромосом составляет 45.
И, наконец, еще один вид патологии – мозаичный синдром Дауна. Его особенность проявляется тем, что пресловутая пара хромосом утраивается, но патология затрагивает только часть клеток.




